a segregating population comprising of f2 individuals was developed using 3096a (female parent) as sterile and 866r (male parent) as restorer, and this study mapped the candidate fertility restoration on 1.35mb of chrd05 and 20 candidate genes were identified for the first time, revealing that there may be differences between lines of 104-7a and gossypium harknessii in fertility restoration genes. moreover, 42 indel markers of the whole genome resequencing were also detected. these results will provide important information for further study of cms restoration genes in cotton.
abstract:
background: cytoplasmic male sterility (cms) is a maternally inherited trait failing to produce functional pollen. it plays a pivotal role in the exploitation of crop heterosis. the specific locus amplified fragment sequencing (slaf-seq) as a high-resolution strategy for the identification of new snps on a large-scale is gradually applied for functional gene mining. the current study combined the bulked segregant analysis (bsa) with slaf-seq to identify the candidate genes associated with fertility restorer gene (rf) in cms cotton.
methods: illumina sequencing systematically investigated the parents. a segregating population comprising of 30 30 f2 individuals was developed using 3096a (female parent) as sterile and 866r (male parent) as a restorer. the original data obtained by dual-index sequencing were analyzed to obtain the reads of each sample that were compared to the reference genome in order to identify the slaf tag with a polymorphism in parent lines and the snp with read-associated coverage. based on slaf tags, snp-index analysis, euclidean distance (ed) correlation analysis, and whole genome resequencing, the hot regions were annotated.
results: a total of 165,007 high-quality slaf tags, with an average depth of 47.90× in the parents and 50.78× in f2 individuals, were sequenced. in addition, a total of 137,741 snps were detected: 113,311 and 98,861 snps in the male and female parent, respectively. a correlation analysis by snp-index and ed initially located the candidate gene on 1.35 mb of chrd05, and 20 candidate genes were identified. these genes were involved in genetic variations, single base mutations, insertions, and deletions. moreover, 42 indel markers of the whole genome resequencing were also detected.
conclusions: in this study, associated markers identified by super-bsa could accelerate the study of cms in cotton, and as well as in other crops. some of the 20 genes’ preliminary characteristics provided useful information for further studies on cms crops.
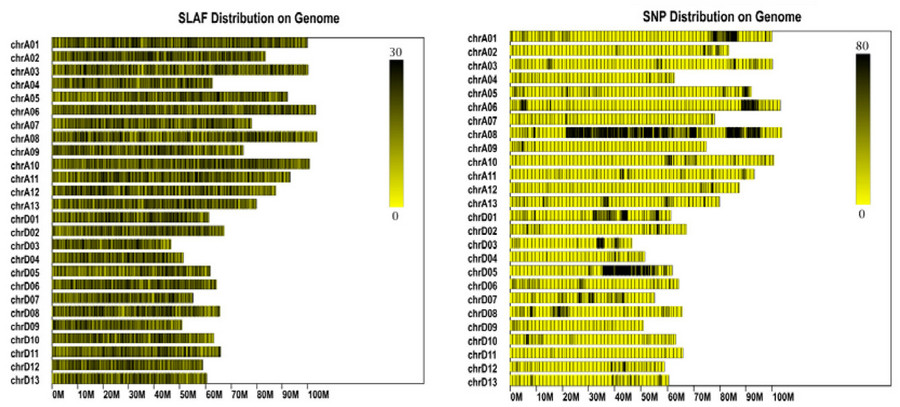
fig. 1 slaf distribution and snp markers on chromosome. note: the abscissa is the length of the chromosome. each yellow band represents a chromosome. the genome is divided by every 1mbp. the more the number of slaf tags in each window, the deeper the color and lesser the number of slaf tags, the lighter the color. the darker area in the figure is the area where the slaf tags are centrally distributed. the left panel shows the distribution of the slaf tag, and the right panel is the distribution of snp.